Background
Primary CNS tumors are the most common pediatric solid cancer, and account for 23% of malignancy in children under 15 years of age, second only to leukemia. The incidence of newly diagnosed brain tumors in the pediatric population is 3.3 cases per 100,000 children, with 3,000 children diagnosed annually. Brain tumors are also the leading cause of cancer death in children. Pediatric brain tumors differ from brain tumors in adults. In adults, supratentorial high grade glioma (WHO-Grade IV, also known as glioblastoma multiforme, is the most common primary brain tumor. In contrast, over 60% of pediatric brain tumors are infratentorial, occurring below the tentorium in the cerebellum or brainstem, and pediatric gliomas are more often low-grade (WHO-Grade I-II). In addition, brainstem gliomas are exceeding rare in adults, while 10-15% of pediatric brain tumors are localized to the brainstem. Over 50% of pediatric brainstem tumors are brainstem gliomas.
Brainstem Gliomas (BSGs) and Diffuse Intrinsic Pontine Gliomas (DIPGs)
Brainstem Gliomas (BSGs) affect 200-300 children in the United State each year, and are the leading cause of brain tumor death in children. BSG’s are classified into four categories on the basis of anatomic location and radiographic appearance: Diffuse, focal intrinsic, focal exophytic, and cervicomedullary. Of these, the most common form is Diffuse intrinsic Pontine Glioma (DIPG), accounting for 80% of all brainstem gliomas.
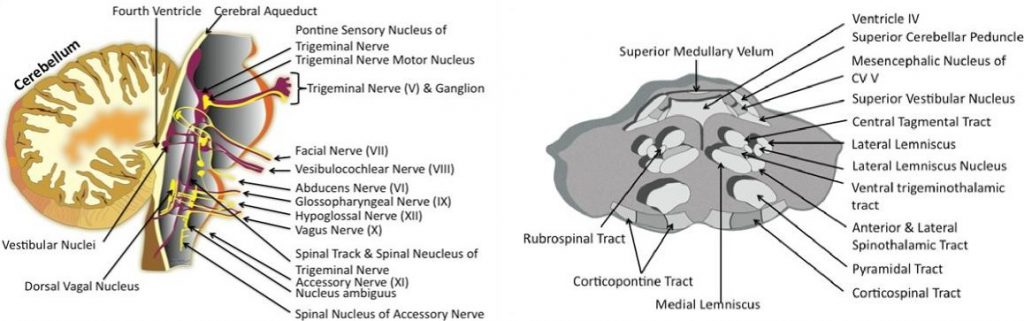
The brainstem is composed of three segments: the midbrain, pons, and medulla oblongata. These structures are made up of densely compact neurologic tissue. Neurons are the specialized cells of the nervous system that transmit electrical and chemical signals to convey information between the brain and the body. Neurons are composed of a cell body, which is the control center of the cell, long processes called axons that extend from the cell body to carry signals to other neurons, and short processes called dendrites which also contribute to the network of connections between neurons. The brainstem is the site of fragile clusters of critical neuron cell bodies, called nuclei, which control respiration, heart beat and blood pressure, swallowing, face and eye sensation and movement, balance, and other important functions (Figure 1). The bulk of the brainstem is made of bundles of axons known as tracts, which carry information back and forth between cells throughout all regions of the brain and the spinal cord to allow coordination of organ function, body movement and sensation (Figure 2).
Tumor cells of a developing DIPG gradually compress crucial nuclei and tracts within the pons. As the tumor enlarges, it causes symptoms due to impaired function of neurons arising in or running through the pons that carry information to and from the cerebellum, cerebral cortex, spinal cord, and cranial nerves. The most common symptoms are a triad of 1) cerebellar deficits such as impaired balance and coordination, 2) long tract impairment causing weakness or sensory loss of the extremities or trunk, and 3) paresis of cranial nerves VI and VII, which facilitate outward movement of the eye and face, respectively. Headaches, altered level of consciousness and other cranial nerve deficits due to obstruction of cerebral spinal fluid flow due to tumor impingement on the ventricular system of the brain, are also not uncommon.
Once a clinician suspects a brainstem abnormality, the radiologic study of choice is Magnetic Resonance Imaging (MRI). DIPG has a typical radiographic appearance on MRI, with diffuse enlargement and tumor infiltration of the pons, and minimal to no enhancement with contrast administration. Most DIPGs are >2 cm at the time of diagnosis, and have a high likelihood of leptomeningeal craniospinal dissemination. DIPG typically exhibits rapid clinical onset and progression, and has a 90% mortality rae within 18 months of presentation. There is a peak incidence of 6-9 years of age, with a mean survival of 10-12 months.
Treatments
There is currently no effective treatments for DIPG. Due to the anatomic location and infiltrative nature of DIPG, these tumors are not amenable to surgical resection. Diagnosis is based on characteristic appearance on MR Imaging, so diagnostic biopsy is rarely performed except in rare cases where diagnosis is uncertain. As a result, tumor tissue for pathologic evaluation and molecular study is rare, and little is known about the molecular biology of DIPG compared to other gliomas, making treatment more challenging. Conventional radiation therapy (RT) is typically used for palliation, providing a transient improvement in neurologic function and progression free survival benefit up to one year with minimal side effects. However, median onset of disease progression following RT is less than 6 months, with no improvement in overall survival. Prolonged survival up to 24 months has been observed in less than 10% of patients. Clinical trials to date investigating radiation fractionation regimens, adjuvant and neoadjuvant chemotherapeutics have not led to advancement in the treatment paradigm of DIPG. Hyperfractionated RT provides no survival benefit compared to conventional or hypofractionated RT, with increased toxicity. Likewise, clinical trials of
single and multi-agent regimens of conventional, myeloblative and radiation sensitizatizing chemotherapeutics have been conducted with no effect on outcome. Varied timing of administration with radiation has also shown no appreciable change in median survival or survival benefit.
Clinical Trials
New clinical trials are currently underway, investigating novel medications and therapeutic techniques in an effort to better treat patients with DIPG. A number of centers, including St. Jude’s Children’s Research Hospital and National Cancer Institute are investigating new combinations and dosages of chemotherapeutic agents in conjunction with RT for patients with DIPG. National Institute of Neurological Disorders and Stroke, parte of National Institutes of Health, is conducting a trial of an experimental drug, IL13-PE38QQR, for the treatment of DIPG and other high grade gliomas using a technique termed convection enhanced delivery (CED). CED involves inserting a catheter into the brain and directly administrating the medication to the tumor itself. Researchers at Stanford University, along with the National Institutes of Health, are investigating the effectiveness of a post-radiation vaccine against Epidermal Growth Factor Receptor (EGFR), a molecule thought to be implicated in the development of high grade gliomas. A large team of clinicians from a variety of pediatric medical centers are also participating in a new clinical trial where surgical biopsy of DIPG tissue will be taken prior to RT administration and inspected by researchers for particular molecular characteristics to help determine adjuvant chemotherapy. By evaluating tumor response to these new treatment techniques, these trials could potentially reveal new and more effective treatment options in the battle against DIPG
Ongoing Research
Unfortunately, the lack of advances in the treatment for DIPG is due in great part to the lack of adequate characterization of the biology of this tumor. As described above, the diffuse nature and location of DIPG precludes surgical removal, and biopsy is not routinely performed for diagnosis, so there is a paucity of fresh tumor tissue for researchers to study. Rare biopsy and post mortem specimens have revealed histological characteristics typical of high grade glioma (WHO-III or IV), including microvascular proliferation, cellular necrosis and the presence of mitotic figures. However further research has shown that pediatric high grade gliomas are biologically distinct from adult high grade gliomas. For example, studies have revealed that signaling pathways giving rise to pediatric gliomas are stimulated by platelet-derived growth factor (PDGF), in contrast to adults, where epidermal growth factor receptor (EGFR) is the predominant signaling abnormality. However, recent data suggest that DIPG and BSG exhibit molecular behavior distinct from supratentorial high grade pediatric gliomas, which could account for the lack of response to treatments that are historically more effective for other tumors.
Researchers are currently developing innovative studies using state-of-the-art technology to better clarify the role of PDGF and other signaling pathways to understand the biology of DIPG. Recent technological advances have made rapid profiling of tumor DNA and RNA arrays possible, revealing differences in gene expression that could help explain tumor formation and be used to develop drug targets. Because DIPG tumor tissue is rare, researchers are also evaluating blood and cerebrospinal fluid samples from patients with DIPG for molecular analysis, as well as cell cultures grown from donated DIPG samples as a medium for investigation. Scientists have also been able to develop reliable transgenic animal models of DIPG, which can facilitate characterization of tumor cell behavior and the investigation therapeutic targets.
Proteomic analysis has also emerged as a valuable tool, enabling rapid, comprehensive identification of the protein profile of a tumor, revealing differentially expressed proteins compared with normal brain tissue. This information can be valuable for elucidating pathways of tumor formation, and may reveal potential tumor biomarkers that can be useful for clinical diagnosis and act as targets for novel therapies. We have previously reported a complete protein profiling of DIPG using formalin fixed postmortem specimens. However, there is a dire need for collection and analysis of fresh frozen postmortem tumor specimens, because fresh specimens are ideal for molecular analysis including DNA, RNA, microRNA and protein profiling. Based on the limited data that is available, DIPG and BSG seem to arise due to multiple molecular abnormalities, and preliminary results suggest there are subtypes of DIPG and BSG with different causative pathways. Thus, access to a greater number of tumor specimens will increase the changes of detecting tumor subtypes, which can be further characterized in the hopes of eventually designing subtype-specific treatments. We and other scientists have recently launched clinical studies for collecting fresh tumor samples, including biopsy tissue and postmortem specimens, with the aim of analyzing the molecular make up of the disease and understanding its molecular pathways (clinicaltrials.gov).
Researchers hope to use the information acquired through these and other studies to develop new, innovative tools in the battle against DIPG, such as biologic constructs that specifically target and tumor cells to make them more sensitive to traditional radiation or chemotherapy, or molecules that shut down tumor formation pathways early in development while sparing normal brain tissue. Pediatric BSG formation is likely the result of a complex interaction of aberrant development and tumorigenic pathways, giving to a variety of biologically distinct tumor subtypes, including DIPG. Studies that aim to characterize the molecular basis of this disease provide the best chance of achieving a better outcome for children affected by this devastating tumor. Those who have devoted work to this end are determined to advancing the understanding of DIPG and finding more effective therapies to provide children suffering from DIPG a better prognosis, and ultimately one day, a cure.
